Resample Univariate or Multivariate Dimorphism
resampleSSD.RdFunction to generate a set of resampled dimorphism estimates for a univariate or multivariate sample.
Called by SSDtest and bootdimorph.
Usage
resampleSSD(
x,
struc = NULL,
compsex = NULL,
npersample = NA,
nResamp = 10000,
exact = F,
limit = 5e+05,
matchvars = F,
replace = F,
datastruc = NULL,
methsMulti = NULL,
methsUni = c("MMR", "BDI"),
sex.female = 1,
center = "geomean",
templatevar = NULL,
na.rm = T,
ncorrection = F,
details = F
)Arguments
- x
A matrix or data frame of measurements from a comparative sample, with rows corresponding to individual specimens and columns corresponding to size variables. Sex data should not be included. Should not include
NAs; resampling addresses will not be affected, but these addresses may generate errors when used by other functions.- struc
Structure information for the data set being compared against, typically from one or more fossil samples with missing data.
strucmust be either (1) a matrix or dataframe of measurements (which can includeNAs) or (2) a vector of integer sample sizes for each variable.- compsex
A vector indicating sex for the individuals in
x. Sex information is not included in any calculations in this function but will be included as a list element in the returned object. Defaults toNULL.- npersample
Integer specifying the sample size of resampled datasets. Defaults to
NA. If set toNA, the sample size of resampled datasets is set equal to the sample size ofx.- nResamp
Integer specifying the number of resampling iterations to calculate addresses for if Monte Carlo sampling is used.
- exact
Logical scalar specifying whether to sample all unique combinations of sample size
npersamplefromx. Defaults toFALSE. If set toFALSE, or if set toTRUEand the number of unique combinations exceedslimit, then Monte Carlo sampling is used instead.- limit
Integer setting the upper limit on the number of unique combinations allowable for exact resampling. If exact resampling would produce more resampled datasets than this number, Monte Carlo resampling is used instead. Defaults to 500,000.
- matchvars
Logical scalar specifying whether to compare the variable names in
comparativeandstrucand pare them both down to the set of shared variables. IfFALSEand variable names differ then an error will be returned. Defaults toFALSE.- replace
Logical scalar passed to
samplespecifying whether or not to sample with replacement. Defaults toFALSE.- datastruc
If multivariate data are used, this is a character string specifiying whether to incorporate the missing data structure into dimorphism estimates (
"missing"), whether to downsample to the missing data sample size but keep all metric data for the comparative sample ("complete"), or to perform both types of resampling separately ("both"). Defaults to"missing", and ignored if only univariate data are provided.- methsMulti
A character string specifying the multivariate method used to calculate or estimate dimorphism. Note that regardless of the value of this argument, multivariate estimation procedures will only be carried out if
xis a multivariate dataset. Seedimorphfor options.- methsUni
A character string specifying the univariate method used to calculate or estimate dimorphism. See
dimorphfor options.- sex.female
An integer scalar (1 or 2) specifying which level of
sexcorresponds to female. Ignored ifsexisNULL. Defaults to 1.- center
A character string specifying the method used to calculate a mean, either
"geomean"(default) which uses the geometric mean, or"mean"which uses the arithmetic mean. More broadly,"geomean"indicates analyses are conducted in logarithmic data space and"mean"indicates analyses are conducted in raw data space. Some methods can only be applied in one domain or the other:"CV"and"CVsex"are always calculated in raw data space andcenterwill be set to"mean"for these methods regardless of the value set by the user;"MoM","sdlog", and"sdlogsex"are always calculated in logarithmic data space andcenterwill be set to"geomean"for these methods regardless of the value set by the user.- templatevar
A character object or integer value specifying the name or column number of the variable in
xto be estimated using the template method. Ignored if template method is not used. Defaults toNULL.- na.rm
A logical scalar indicating whether NA values should be stripped before the computation proceeds. Defaults to
TRUE.- ncorrection
A logical scalar indicating whether to apply Sokal and Braumann's (1980) size correction factor to CV estimates. Defaults to
FALSE.- details
A logical scalar indicating whether variable name and specimen names should be retained (if available) as attributes in the output object. Defaults to
FALSE.
Value
A list of class dimorphResampledUni or dimorphResampledMulti containing a dataframe
with resampled dimorphism estimates and a dimorphAds object containg resampled addresses produced
by getsampleaddresses. Plotting this object produces violin plots for
all resampled distributions.
Examples
## Univariate
data(apelimbart)
gor <- apelimbart[apelimbart$Species=="Gorilla gorilla",]
# this is effectively a bootstrap, although see 'bootdimorph'
gorSSD <- resampleSSD(gor[,"FHSI", drop=FALSE], methsUni=c("SSD", "MMR", "BDI"),
compsex=gor$Sex, nResamp=100, replace=TRUE)
gorSSD
#> dimorphResampledUni Object
#>
#> Comparative data set:
#> number of specimens: 47 female, 47 male
#> number of variables: 1
#> variable name: FHSI
#> SSD estimate methods (univariate):
#> SSD, MMR, BDI
#> Centering algorithms:
#> geometric mean
#> Number of unique combinations of univariate method and centering algorithm: 3
#>
#> Resampling data structure:
#> type of resampling: Monte Carlo
#> number of resampled data sets: 100
#> number of individuals in each resampled data set: 94
#> subsamples sampled WITH replacement
#> other resampling parameters:
#> sex data present
#> ratio variables (if present): natural log of ratio
#> matchvars = FALSE
#> na.rm = TRUE
plot(gorSSD)
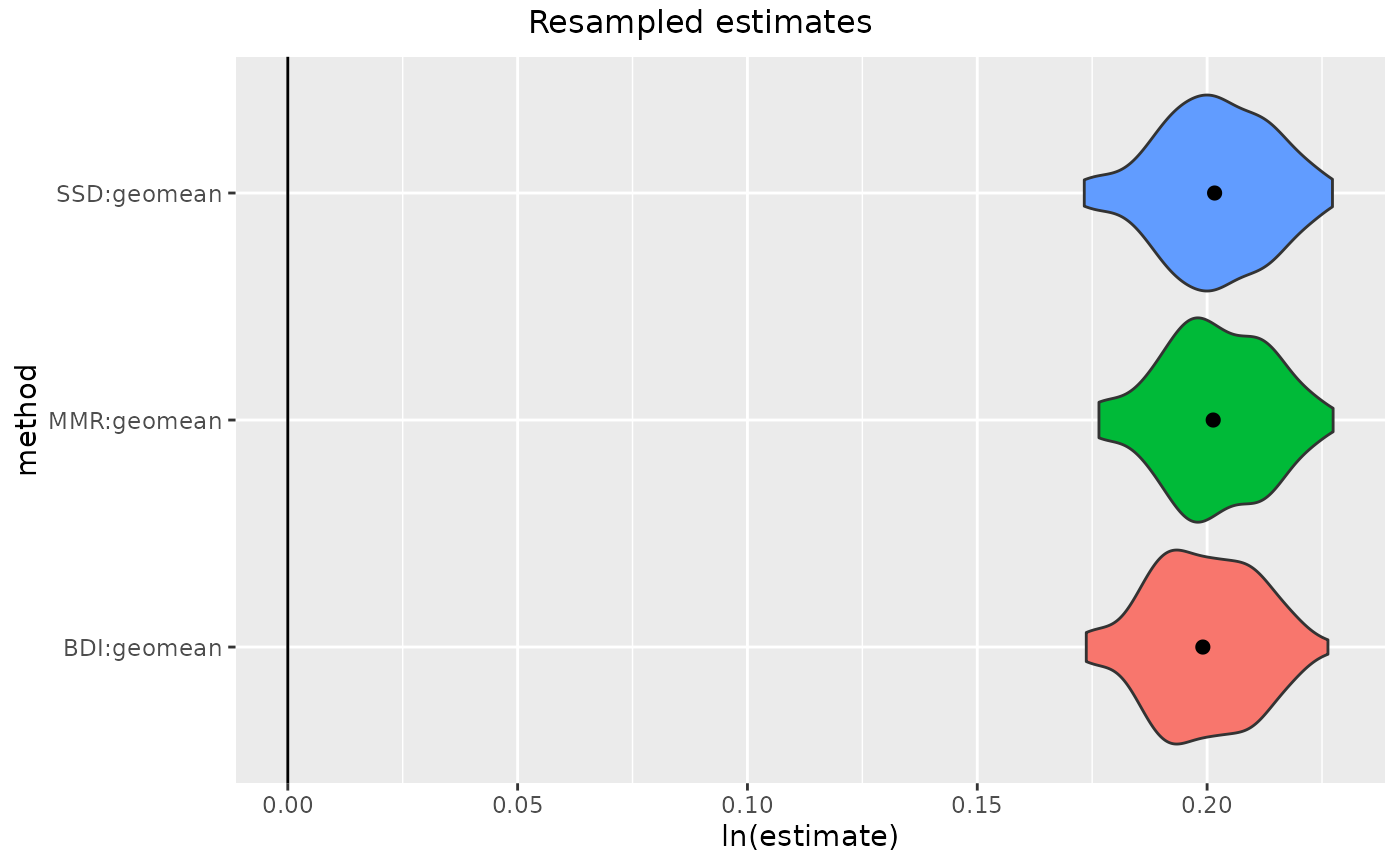 # now downsample to fossil sample size and sample without replacement
SSDvars <- c("HHMaj")
gorSSD1 <- resampleSSD(x=apelimbart[apelimbart$Species=="Gorilla gorilla", SSDvars, drop=FALSE],
struc=fauxil[fauxil$Species=="Fauxil sp. 1", SSDvars, drop=FALSE],
compsex=apelimbart[apelimbart$Species=="Gorilla gorilla", "Sex"],
exact=TRUE, matchvars=TRUE, replace=FALSE, methsUni=c("SSD", "MMR", "BDI"))
#> Warning: The number of possible combinations (54891018) exceeds the user-specified limit. Monte Carlo sampling will be used.
gorSSD1
#> dimorphResampledUni Object
#>
#> Comparative data set:
#> number of specimens: 47 female, 47 male
#> number of variables: 1
#> variable name: HHMaj
#> SSD estimate methods (univariate):
#> SSD, MMR, BDI
#> Centering algorithms:
#> geometric mean
#> Number of unique combinations of univariate method and centering algorithm: 3
#>
#> Resampling data structure:
#> type of resampling: Monte Carlo
#> number of resampled data sets: 10000
#> number of individuals in each resampled data set: 5
#> subsamples sampled WITHOUT replacement
#> other resampling parameters:
#> sex data present
#> ratio variables (if present): natural log of ratio
#> matchvars = TRUE
#> na.rm = TRUE
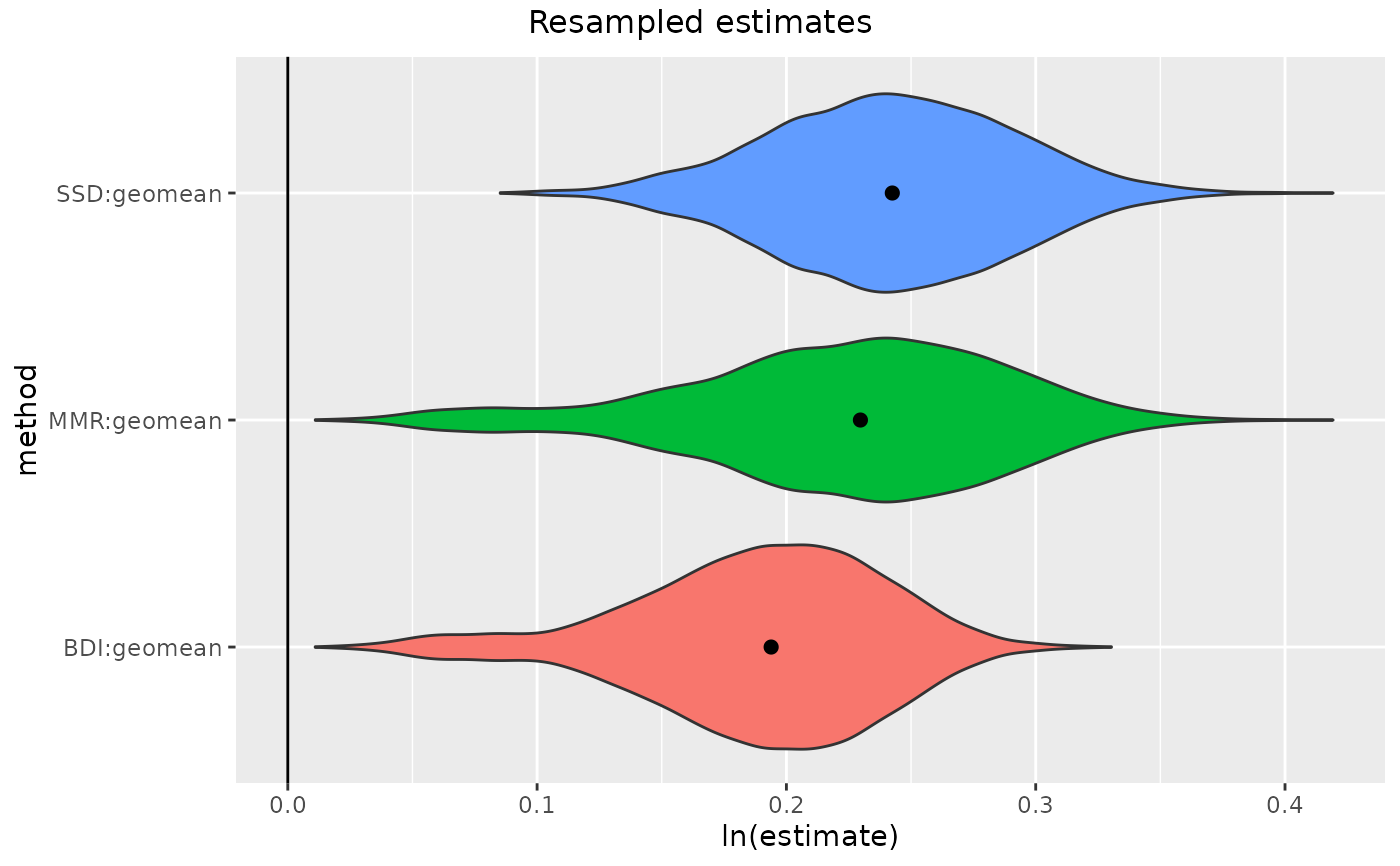
plot(gorSSD1)
# now downsample to fossil sample size and sample without replacement
SSDvars <- c("HHMaj")
gorSSD1 <- resampleSSD(x=apelimbart[apelimbart$Species=="Gorilla gorilla", SSDvars, drop=FALSE],
struc=fauxil[fauxil$Species=="Fauxil sp. 1", SSDvars, drop=FALSE],
compsex=apelimbart[apelimbart$Species=="Gorilla gorilla", "Sex"],
exact=TRUE, matchvars=TRUE, replace=FALSE, methsUni=c("SSD", "MMR", "BDI"))
#> Warning: The number of possible combinations (54891018) exceeds the user-specified limit. Monte Carlo sampling will be used.
gorSSD1
#> dimorphResampledUni Object
#>
#> Comparative data set:
#> number of specimens: 47 female, 47 male
#> number of variables: 1
#> variable name: HHMaj
#> SSD estimate methods (univariate):
#> SSD, MMR, BDI
#> Centering algorithms:
#> geometric mean
#> Number of unique combinations of univariate method and centering algorithm: 3
#>
#> Resampling data structure:
#> type of resampling: Monte Carlo
#> number of resampled data sets: 10000
#> number of individuals in each resampled data set: 5
#> subsamples sampled WITHOUT replacement
#> other resampling parameters:
#> sex data present
#> ratio variables (if present): natural log of ratio
#> matchvars = TRUE
#> na.rm = TRUE
plot(gorSSD1)
 # or run 'getsampleaddresses' first
addressesUni <- getsampleaddresses(
comparative=apelimbart[apelimbart$Species=="Gorilla gorilla", SSDvars, drop=FALSE],
struc=fauxil[fauxil$Species=="Fauxil sp. 1", SSDvars, drop=FALSE],
compsex=apelimbart[apelimbart$Species=="Gorilla gorilla", "Sex"],
exact=TRUE, matchvars=TRUE, replace=FALSE)
#> Warning: The number of possible combinations (54891018) exceeds the user-specified limit. Monte Carlo sampling will be used.
gorSSD2 <- resampleSSD(addressesUni, methsUni=c("SSD", "MMR", "BDI"))
gorSSD2
#> dimorphResampledUni Object
#>
#> Comparative data set:
#> number of specimens: 47 female, 47 male
#> number of variables: 1
#> variable name: HHMaj
#> SSD estimate methods (univariate):
#> SSD, MMR, BDI
#> Centering algorithms:
#> geometric mean
#> Number of unique combinations of univariate method and centering algorithm: 3
#>
#> Resampling data structure:
#> type of resampling: Monte Carlo
#> number of resampled data sets: 10000
#> number of individuals in each resampled data set: 5
#> subsamples sampled WITHOUT replacement
#> other resampling parameters:
#> sex data present
#> ratio variables (if present): natural log of ratio
#> matchvars = TRUE
#> na.rm = TRUE
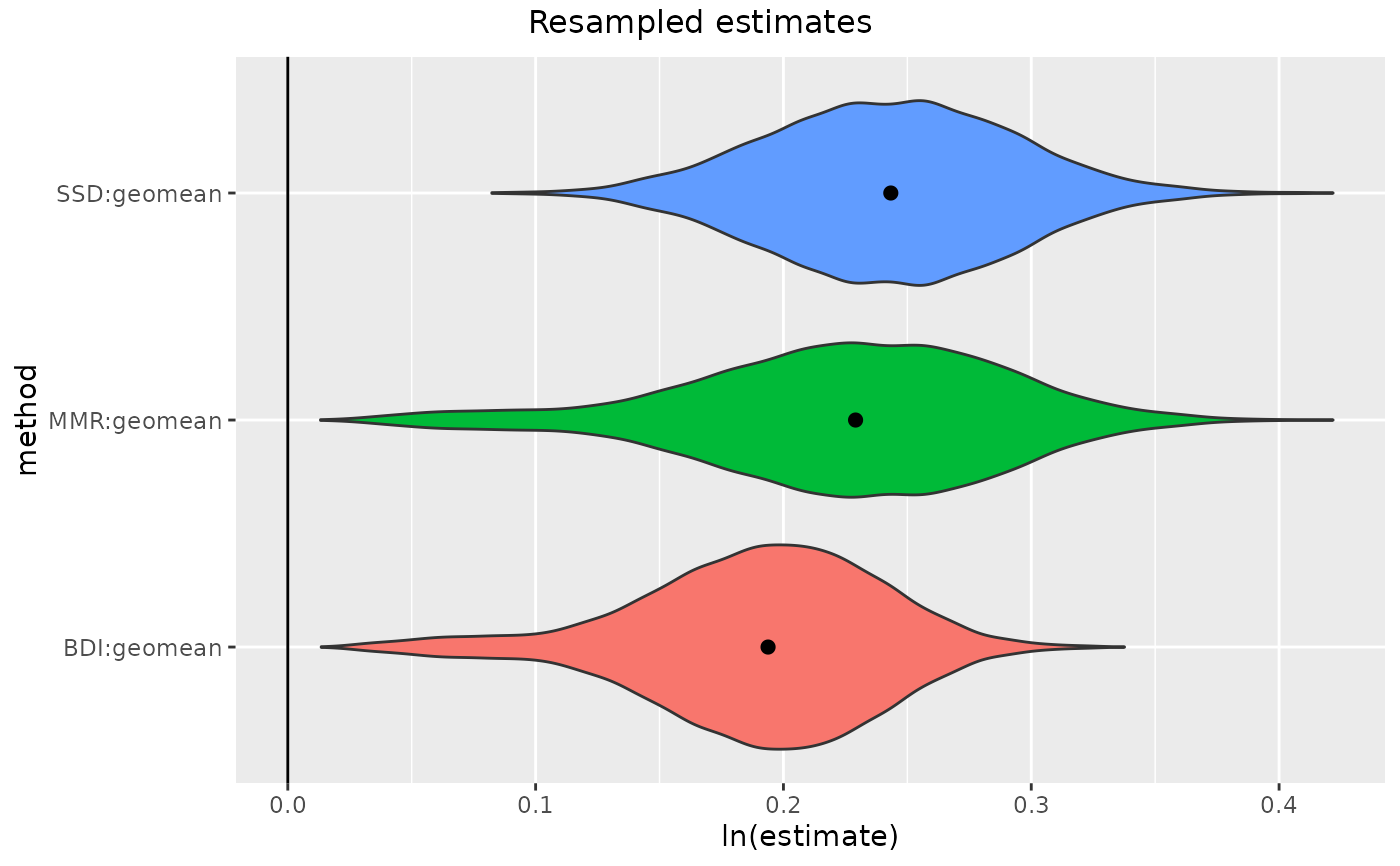
plot(gorSSD2)
# or run 'getsampleaddresses' first
addressesUni <- getsampleaddresses(
comparative=apelimbart[apelimbart$Species=="Gorilla gorilla", SSDvars, drop=FALSE],
struc=fauxil[fauxil$Species=="Fauxil sp. 1", SSDvars, drop=FALSE],
compsex=apelimbart[apelimbart$Species=="Gorilla gorilla", "Sex"],
exact=TRUE, matchvars=TRUE, replace=FALSE)
#> Warning: The number of possible combinations (54891018) exceeds the user-specified limit. Monte Carlo sampling will be used.
gorSSD2 <- resampleSSD(addressesUni, methsUni=c("SSD", "MMR", "BDI"))
gorSSD2
#> dimorphResampledUni Object
#>
#> Comparative data set:
#> number of specimens: 47 female, 47 male
#> number of variables: 1
#> variable name: HHMaj
#> SSD estimate methods (univariate):
#> SSD, MMR, BDI
#> Centering algorithms:
#> geometric mean
#> Number of unique combinations of univariate method and centering algorithm: 3
#>
#> Resampling data structure:
#> type of resampling: Monte Carlo
#> number of resampled data sets: 10000
#> number of individuals in each resampled data set: 5
#> subsamples sampled WITHOUT replacement
#> other resampling parameters:
#> sex data present
#> ratio variables (if present): natural log of ratio
#> matchvars = TRUE
#> na.rm = TRUE
plot(gorSSD2)
 ## Multivariate
SSDvars <- c("HHMaj","RHMaj","FHSI","TPML")
gorSSDmulti1 <- resampleSSD(x=apelimbart[apelimbart$Species=="Gorilla gorilla", SSDvars],
struc=fauxil[fauxil$Species=="Fauxil sp. 1", SSDvars],
compsex=apelimbart[apelimbart$Species=="Gorilla gorilla", "Sex"],
nResamp=100,
datastruc="complete",
methsMulti = c("GMM"),
methsUni = c("SSD", "MMR", "BDI"),
matchvars=TRUE,
replace=FALSE)
gorSSDmulti1
#> dimorphResampledMulti Object
#>
#> Comparative data set:
#> number of specimens: 47 female, 47 male
#> number of variables: 4
#> variable names: HHMaj, RHMaj, FHSI, TPML
#> SSD estimate methods (univariate):
#> SSD, MMR, BDI
#> SSD estimate methods (multivariate):
#> GMM
#> Centering algorithms:
#> geometric mean
#> Multivariate sampling with complete or missing data:
#> complete
#> Number of unique combinations of univariate method, multivariate method,
#> centering algorithm, and complete or missing data structure: 3
#>
#> Resampling data structure:
#> type of resampling: Monte Carlo
#> number of resampled data sets: 100
#> number of individuals in each resampled data set: 10
#> proportion of missing data in resampling structure: 0.625
#> subsamples sampled WITHOUT replacement
#> other resampling parameters:
#> sex data present
#> ratio variables (if present): natural log of ratio
#> matchvars = TRUE
#> na.rm = TRUE
plot(gorSSDmulti1)
## Multivariate
SSDvars <- c("HHMaj","RHMaj","FHSI","TPML")
gorSSDmulti1 <- resampleSSD(x=apelimbart[apelimbart$Species=="Gorilla gorilla", SSDvars],
struc=fauxil[fauxil$Species=="Fauxil sp. 1", SSDvars],
compsex=apelimbart[apelimbart$Species=="Gorilla gorilla", "Sex"],
nResamp=100,
datastruc="complete",
methsMulti = c("GMM"),
methsUni = c("SSD", "MMR", "BDI"),
matchvars=TRUE,
replace=FALSE)
gorSSDmulti1
#> dimorphResampledMulti Object
#>
#> Comparative data set:
#> number of specimens: 47 female, 47 male
#> number of variables: 4
#> variable names: HHMaj, RHMaj, FHSI, TPML
#> SSD estimate methods (univariate):
#> SSD, MMR, BDI
#> SSD estimate methods (multivariate):
#> GMM
#> Centering algorithms:
#> geometric mean
#> Multivariate sampling with complete or missing data:
#> complete
#> Number of unique combinations of univariate method, multivariate method,
#> centering algorithm, and complete or missing data structure: 3
#>
#> Resampling data structure:
#> type of resampling: Monte Carlo
#> number of resampled data sets: 100
#> number of individuals in each resampled data set: 10
#> proportion of missing data in resampling structure: 0.625
#> subsamples sampled WITHOUT replacement
#> other resampling parameters:
#> sex data present
#> ratio variables (if present): natural log of ratio
#> matchvars = TRUE
#> na.rm = TRUE
plot(gorSSDmulti1)
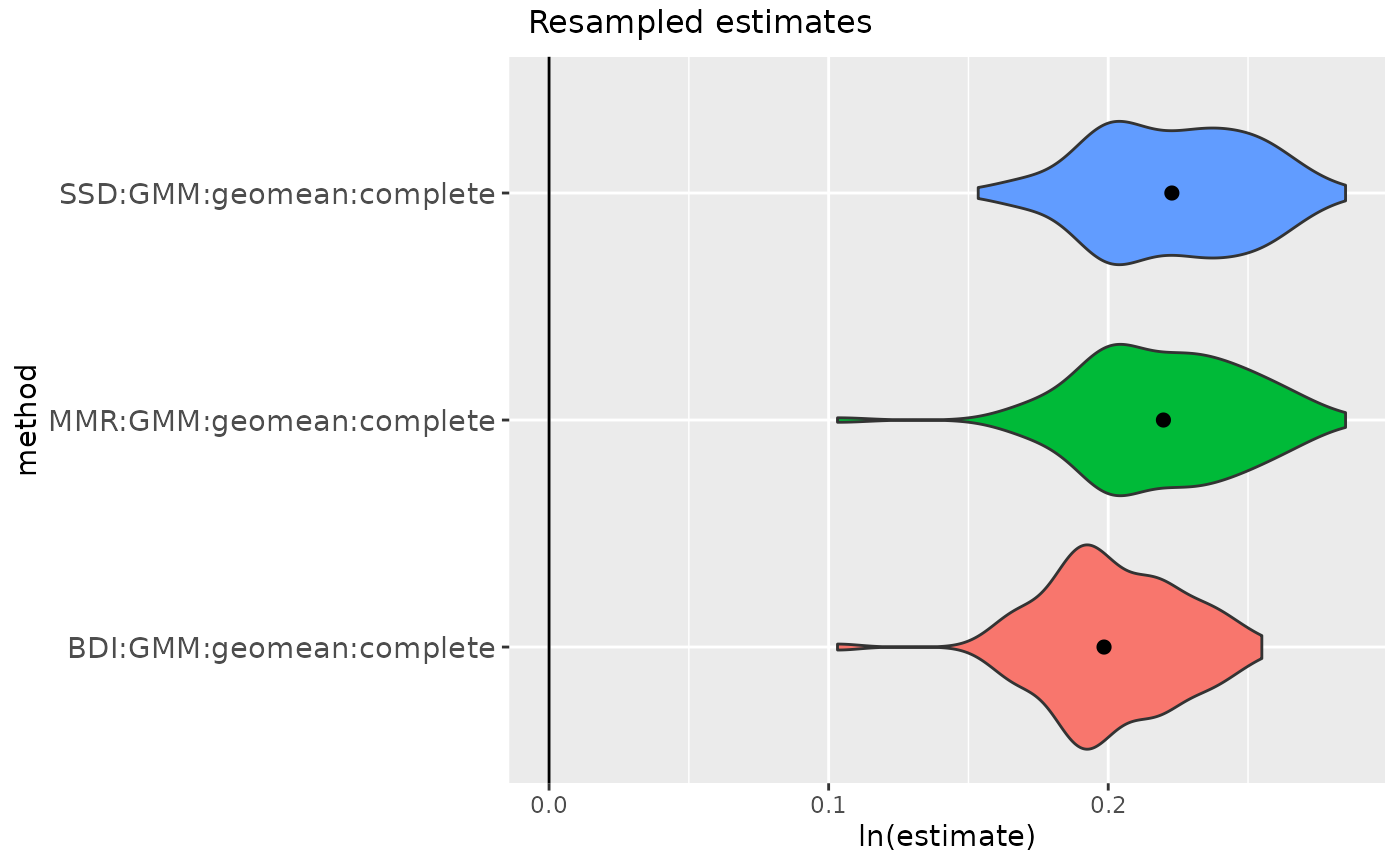 # or run 'getsampleaddresses' first
addresses <- getsampleaddresses(comparative=
apelimbart[apelimbart$Species=="Gorilla gorilla", SSDvars],
struc=fauxil[fauxil$Species=="Fauxil sp. 1", SSDvars],
compsex=apelimbart[apelimbart$Species=="Gorilla gorilla", "Sex"],
nResamp=100, matchvars=TRUE, replace=FALSE)
gorSSDmulti2 <- resampleSSD(x=addresses,
datastruc="complete",
methsMulti = c("GMM"),
methsUni = c("SSD", "MMR", "BDI"))
#> Warning: SSD can not be calculated when only one sex is present.
#> Warning: SSD can not be calculated when only one sex is present.
#> Warning: SSD can not be calculated when only one sex is present.
#> Warning: SSD can not be calculated when only one sex is present.
#> Warning: The following variable(s) were removed because they generated
#> NA estimates:
#> HHMaj, RHMaj, FHSI, TPML
#> Warning: The following specimen(s) were removed because they did not include
#> at least one measurement after variables were removed:
#> PCM Gg-M174, PCM Gg-M902, PCM Gg-M138, CMNH HTB 1856, PCM Gg-C1-098, PCM Gg-M095, RBINS 33238, CMNH HTB 1854, PCM Gg-M096, PCM Gg-Z6-33
#> Warning: argument is not numeric: returning NA
gorSSDmulti2
#> dimorphResampledMulti Object
#>
#> Comparative data set:
#> number of specimens: 47 female, 47 male
#> number of variables: 4
#> variable names: HHMaj, RHMaj, FHSI, TPML
#> SSD estimate methods (univariate):
#> SSD, MMR, BDI
#> SSD estimate methods (multivariate):
#> GMM
#> Centering algorithms:
#> geometric mean
#> Multivariate sampling with complete or missing data:
#> complete
#> Number of unique combinations of univariate method, multivariate method,
#> centering algorithm, and complete or missing data structure: 3
#>
#> Resampling data structure:
#> type of resampling: Monte Carlo
#> number of resampled data sets: 100
#> number of individuals in each resampled data set: 10
#> proportion of missing data in resampling structure: 0.625
#> subsamples sampled WITHOUT replacement
#> other resampling parameters:
#> sex data present
#> ratio variables (if present): natural log of ratio
#> matchvars = TRUE
#> na.rm = TRUE
plot(gorSSDmulti2)
# or run 'getsampleaddresses' first
addresses <- getsampleaddresses(comparative=
apelimbart[apelimbart$Species=="Gorilla gorilla", SSDvars],
struc=fauxil[fauxil$Species=="Fauxil sp. 1", SSDvars],
compsex=apelimbart[apelimbart$Species=="Gorilla gorilla", "Sex"],
nResamp=100, matchvars=TRUE, replace=FALSE)
gorSSDmulti2 <- resampleSSD(x=addresses,
datastruc="complete",
methsMulti = c("GMM"),
methsUni = c("SSD", "MMR", "BDI"))
#> Warning: SSD can not be calculated when only one sex is present.
#> Warning: SSD can not be calculated when only one sex is present.
#> Warning: SSD can not be calculated when only one sex is present.
#> Warning: SSD can not be calculated when only one sex is present.
#> Warning: The following variable(s) were removed because they generated
#> NA estimates:
#> HHMaj, RHMaj, FHSI, TPML
#> Warning: The following specimen(s) were removed because they did not include
#> at least one measurement after variables were removed:
#> PCM Gg-M174, PCM Gg-M902, PCM Gg-M138, CMNH HTB 1856, PCM Gg-C1-098, PCM Gg-M095, RBINS 33238, CMNH HTB 1854, PCM Gg-M096, PCM Gg-Z6-33
#> Warning: argument is not numeric: returning NA
gorSSDmulti2
#> dimorphResampledMulti Object
#>
#> Comparative data set:
#> number of specimens: 47 female, 47 male
#> number of variables: 4
#> variable names: HHMaj, RHMaj, FHSI, TPML
#> SSD estimate methods (univariate):
#> SSD, MMR, BDI
#> SSD estimate methods (multivariate):
#> GMM
#> Centering algorithms:
#> geometric mean
#> Multivariate sampling with complete or missing data:
#> complete
#> Number of unique combinations of univariate method, multivariate method,
#> centering algorithm, and complete or missing data structure: 3
#>
#> Resampling data structure:
#> type of resampling: Monte Carlo
#> number of resampled data sets: 100
#> number of individuals in each resampled data set: 10
#> proportion of missing data in resampling structure: 0.625
#> subsamples sampled WITHOUT replacement
#> other resampling parameters:
#> sex data present
#> ratio variables (if present): natural log of ratio
#> matchvars = TRUE
#> na.rm = TRUE
plot(gorSSDmulti2)
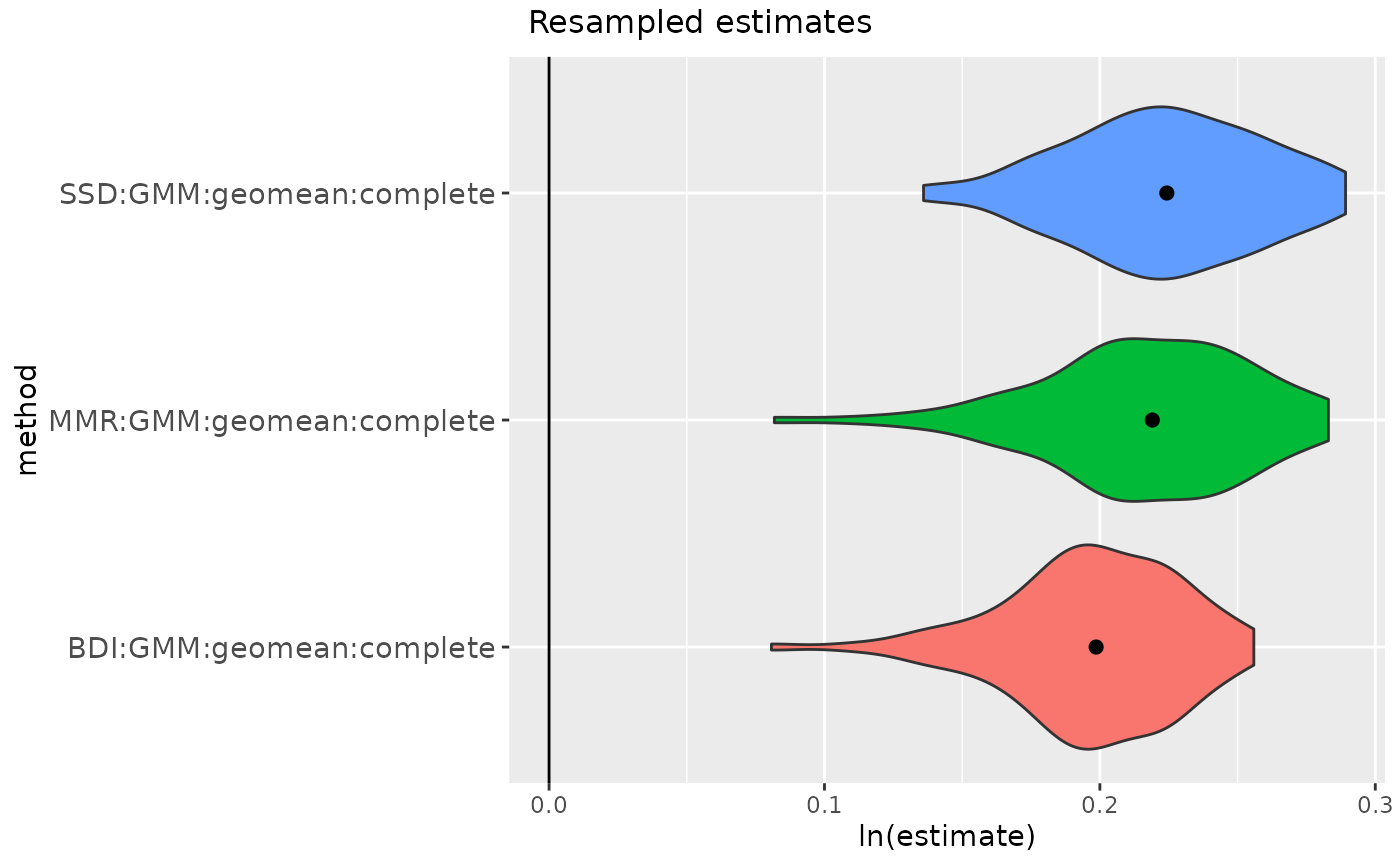 # or run with missing data (exclude 'SSD' because some variables won't have both sexes)
addresses <- getsampleaddresses(comparative=
apelimbart[apelimbart$Species=="Gorilla gorilla", SSDvars],
struc=fauxil[fauxil$Species=="Fauxil sp. 1", SSDvars],
compsex=apelimbart[apelimbart$Species=="Gorilla gorilla", "Sex"],
nResamp=100, matchvars=TRUE, replace=FALSE)
gorSSDmulti3 <- resampleSSD(x=addresses,
datastruc="both",
methsMulti = c("GMM"),
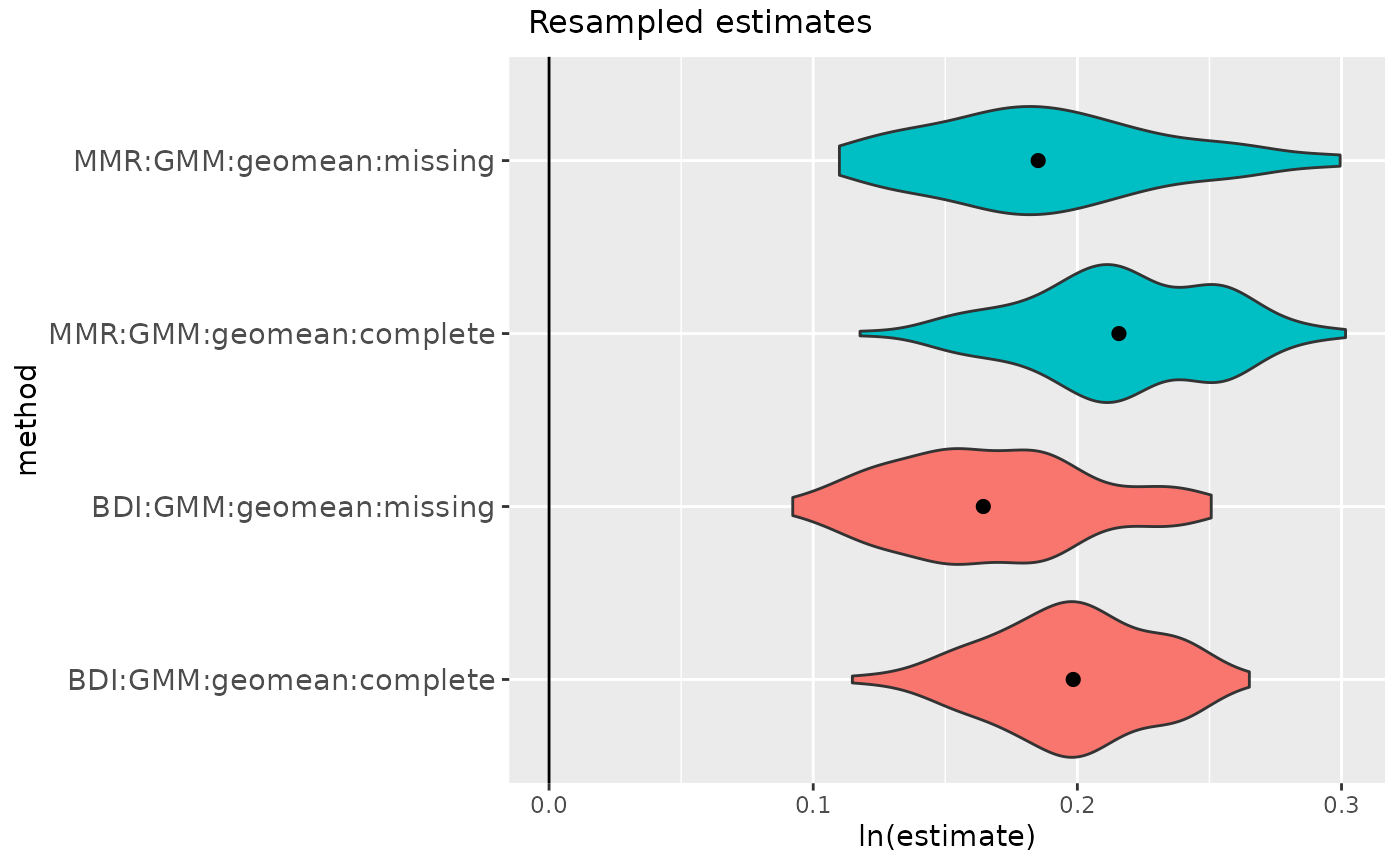
methsUni = c("MMR", "BDI"))
gorSSDmulti3
#> dimorphResampledMulti Object
#>
#> Comparative data set:
#> number of specimens: 47 female, 47 male
#> number of variables: 4
#> variable names: HHMaj, RHMaj, FHSI, TPML
#> SSD estimate methods (univariate):
#> MMR, BDI
#> SSD estimate methods (multivariate):
#> GMM
#> Centering algorithms:
#> geometric mean
#> Multivariate sampling with complete or missing data:
#> complete and missing
#> Number of unique combinations of univariate method, multivariate method,
#> centering algorithm, and complete or missing data structure: 4
#>
#> Resampling data structure:
#> type of resampling: Monte Carlo
#> number of resampled data sets: 100
#> missing data resampling structure:
#> sampling individuals, then imposing missing data pattern
#> number of individuals in each resampled data set: 10
#> proportion of missing data in resampling structure: 0.625
#> subsamples sampled WITHOUT replacement
#> other resampling parameters:
#> sex data present
#> ratio variables (if present): natural log of ratio
#> matchvars = TRUE
#> na.rm = TRUE
plot(gorSSDmulti3)
# or run with missing data (exclude 'SSD' because some variables won't have both sexes)
addresses <- getsampleaddresses(comparative=
apelimbart[apelimbart$Species=="Gorilla gorilla", SSDvars],
struc=fauxil[fauxil$Species=="Fauxil sp. 1", SSDvars],
compsex=apelimbart[apelimbart$Species=="Gorilla gorilla", "Sex"],
nResamp=100, matchvars=TRUE, replace=FALSE)
gorSSDmulti3 <- resampleSSD(x=addresses,
datastruc="both",
methsMulti = c("GMM"),
methsUni = c("MMR", "BDI"))
gorSSDmulti3
#> dimorphResampledMulti Object
#>
#> Comparative data set:
#> number of specimens: 47 female, 47 male
#> number of variables: 4
#> variable names: HHMaj, RHMaj, FHSI, TPML
#> SSD estimate methods (univariate):
#> MMR, BDI
#> SSD estimate methods (multivariate):
#> GMM
#> Centering algorithms:
#> geometric mean
#> Multivariate sampling with complete or missing data:
#> complete and missing
#> Number of unique combinations of univariate method, multivariate method,
#> centering algorithm, and complete or missing data structure: 4
#>
#> Resampling data structure:
#> type of resampling: Monte Carlo
#> number of resampled data sets: 100
#> missing data resampling structure:
#> sampling individuals, then imposing missing data pattern
#> number of individuals in each resampled data set: 10
#> proportion of missing data in resampling structure: 0.625
#> subsamples sampled WITHOUT replacement
#> other resampling parameters:
#> sex data present
#> ratio variables (if present): natural log of ratio
#> matchvars = TRUE
#> na.rm = TRUE
plot(gorSSDmulti3)